Phylometrics
 Phylometrics provides a novel data mining method to screen supplied DNA sequences
and to identify sequences that are of significant phylogenetic interest using powerful
analytical tools. Sequences that are identified as being similar to a number of supplied
sequences may provide key insights into their functional or evolutionary relatedness.
Users require the same basic computer skills as for navigating most internet applications.
Phylometrics provides a novel data mining method to screen supplied DNA sequences
and to identify sequences that are of significant phylogenetic interest using powerful
analytical tools. Sequences that are identified as being similar to a number of supplied
sequences may provide key insights into their functional or evolutionary relatedness.
Users require the same basic computer skills as for navigating most internet applications.
Summary
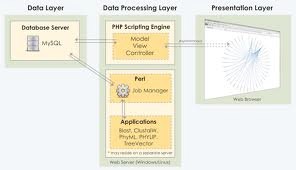
We have developed a web application, Phylometrics, that relies on networks of collective BLASTN results (rather than single BLASTN hits) to facilitate the process of building phylogenetic trees in an automated, high-throughput fashion while offering novel tools to find sequences that are of significant phylogenetic interest with minimal human involvement. The application, which can be installed locally in a laboratory or hosted remotely, utilizes a simple wizard-style format to guide the user through the pipeline without necessitating a background in programming. Furthermore, Phylometrics implements an independent job queuing system that enables users to continue to use the system while jobs are run with little or no degradation in performance.
Background
Comparative sequence analysis of the 16S rRNA gene is frequently used to characterize the microbial diversity of environmental samples. However, sequence similarities do not always imply functional or evolutionary relatedness due to many factors, including unequal rates of change and convergence. Thus, relying on top BLASTN hits for phylogenetic studies may misrepresent the diversity of these constituents. Furthermore, attempts to circumvent this issue by including a large number of BLASTN hits per sequence in one tree to explore their relatedness presents other problems. For instance, the multiple sequence alignment will be poor and computationally costly if not relying on manual alignment, and it may be difficult to derive meaningful relationships from the resulting tree. Analyzing sequence relationship networks within collective BLASTN results, however, reveal sequences that are closely related despite low rank.